
Multiple Myeloma
In our myeloma research we initially concentrated on intra-cellular pro-survival signalling in response to cytotoxic stress and chemotherapy drugs. We identified roles pro-tumoral roles for HO-1 and BTK as well as microRNA125b in MM proliferation. Moreover, we found that in MM, BTK inhibition acts synergistically with proteasome inhibitors (e.g. bortezomib) and IMIDs (e.g. lenalidomide) to kill malignant plasma cells. In relapsed disease, we identified the BTK pathway as important in bortezomib resistance and that BTK inhibition appeared to re-sensitise MM cells to proteasome inhibitor treatment. These studies have subsequently resulted in a number of clinical trials of the BTK inhibitor ibrutinib in MM patients.
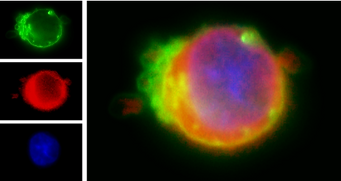
Unpublished data

In parallel with our work in AML we increasingly focussed on the role of the bone marrow microenvironment. We identified isoform specific functions of PI3Kγ and PI3Kδ in the MM microenvironment. We also found a pro-tumoral interaction between MM cells and the BMSC, whereby MM-derived macrophage migratory inhibitory factor (MIF) causes BMSC secretion of IL-6 and IL-8 via BMSC cMYC. Furthermore, we showed that the cMYC inhibitor JQ1 can reduce BMSC secreted IL-6 in vivo, irrespective of tumour burden. These data provide evidence for the clinical evaluation of both MIF and cMYC inhibitors in the treatment of MM.
Marlein et al. Cancer Research 2019
More recently we investigated some of the metabolic processes involved in myeloma disease progression and chemotherapy resistance. We showed that reliance of MM cells on oxidative phosphorylation is mediated by intercellular mitochondrial transfer to MM cells from neighbouring non-malignant BMSC through tumor-derived tunneling nanotubes (TNT). This was shown to be dependent on CD38.

Marlein et al. Cancer Research 2019